ReTrace is a computational method for inferring branching pathways in genome-scale metabolic networks.
ReTrace is also a Python program implementing the method. The software has been licensed under GNU GPL.
Contact: Esa Pitkänen
Esa Pitkänen, Paula Jouhten and Juho Rousu: Inferring branching pathways in genome-scale metabolic networks. BMC Systems Biology 2009, 3:103. Paula Jouhten, Esa Pitkänen, Tiina Pakula, Markku Saloheimo, Merja Penttilä, Hannu Maaheimo: 13C-metabolic flux ratio and novel carbon path analyses confirmed that Trichoderma reesei uses primarily the respirative pathway also on the preferred carbon source glucose. BMC Systems Biology 2009, 3:104.
View changelog.
Provided you have already installed ReTrace successfully and have a local copy of KEGG LIGAND database in directory kegg, you are able to compute branching pathways from metabolite X to metabolite Y by invoking
python retrace.py -d kegg -o results -s X -t Y
The results will be written to the directory results. For instance, to compute pathways between glucose (KEGG LIGAND identifier C00031) and acetyl-CoA (C00024), invoke the command
python retrace.py -d kegg -o results -s C00031 -t C00024
The main result file, written into output/pathways-C00031-to-C00024.html, should resemble the example file you can find here.
| Option | Description | Default value |
|---|---|---|
| -d | KEGG database directory | Required |
| -o | Output directory | Required |
| -s | Source metabolites | Required |
| -t | Target metabolite | Required |
| -a | Traced atom types | |
| -c | Reaction score file | - |
| -e | Atom graph edge weights: (u)niform, (s)cores, (a)toms | Uniform |
| -g | Greedy finish: set k=1 for search levels 2 and beyond | No |
| -i | Report incomplete pathways | No |
| -l | Maximum pathway size | Unbounded |
| -k | Number of shortest paths computed in each step | 50,1 |
| -m | Maximum search depth | 3 |
| -p | Prune atom graph | No pruning |
| -r | Reaction direction constraints file | - |
| -w | Minimum ZO score requirement | 0 |
Command-line options of ReTrace are summarized in above table. The user is required to give the directory (-d) where the local copy of KEGG LIGAND database has been installed, the directory where ReTrace output (-o) is written to, and the source (-s) and target (-t) metabolites.
More than one source metabolites, given as a comma-separated list of KEGG compound identifiers, are admitted (e.g., "C00024,C00026"). If a particular subset of source atoms are of interest, such often is the case with AcCoA, for example, the source atoms can be limited to this subset by giving the atoms as a list after each source atom. The list needs to be separated by a dash (-) from the source metabolite, and atoms in the list by a slash (/). For instance, running ReTrace with the command
python retrace.py -d kegg -o result -s C00024-49/50 -t C00047
would search for paths from AcCoA (C00024) acetyl group carbons (49 and 50 in the atom numbering of KEGG March 2009 version) to Lysine (C00047). This search would adopt the default values for the number of shortest paths computed in each step and the maximum search depth. Particularly only carbon atoms would by traced in search.
Increasing the default number of shortest paths computed with -k results in more pathways being found and a higher computational cost at every search level. For a more fine-tuned control, a comma-separated list of integers can be specified with -k to set k individually for each search level. For instance, a query with -k 50,10,1 would search for 50 shortest paths at the first level, then 10 at the second and 1 at the third and subsequent levels. Option -g provides a quick way of specifying a search where -k option determines the number of paths at the first level but at second and following levels only 1 path is computer per level. This is particularly useful when the focus is on finding different linear connections from sources to target and possible branches can be resolved with any (single) path.
Setting maximum search depth with option -m governs how many branches at maximum appear in result pathways. It should be noted, that with the option -m 1 the method closely corresponds the operation of the ARM method in the sense that it searches for k shortest, unbranching pathways in an atom graph.
As described in the manuscript, ReTrace can take advantage of scores computed for any subset of KEGG reactions. A score file, specified with the -c option, has to contain one reaction-score pair per line, separated by a tab character. Any reaction with no score specified is considered to have a zero score. The -c option should be used in conjunction with the -e option to reweight the atom graph edges by reaction scores. By default edges are assigned uniform weights.
Another weighting option is to give each edge (va, vb) induced by a reaction r the weight 1/ alpha, where alpha is the number of edges in total connecting the metabolites of atoms a and b in reaction r. Therefore, this weighting scheme favors pathways traversing reactions which involve a large number of atoms.
Currently, KEGG data contains mappings for carbon, nitrogen, oxygen and phosphorus atoms. By default, however, ReTrace utilizes only carbon atoms in search. This behavior can be changed with the option -a by giving a comma-separate list of element symbols, for example C,N,P. In general, accurate atom mappings for oxygens are hard to compute because of the typical high degree of symmetry involved. However, when studying nitrogen metabolism, for instance, it is necessary to include also nitrogens in search with this option.
In experiments reported in this study, we found it unnecessary to prune the atom graph induced by KEGG reactions. However, for some purposes, it may be useful to prune the graph to reduce the computation time. To accomplish this, ReTrace supports the -p option which can be supplied an integer n governing the degree of pruning. Specifically, ReTrace prunes the atom graph by considering the total distance of reactions from both sources and target and leaving the n reactions with smallest total distance into the graph and removing the others. Pruning respects the reweighting scheme chosen with the option -e.
If available, ReTrace is able to incorporate constraints to reaction directions in search. This is done via the option -r by supplying the file containing KEGG reaction identifiers and direction constraint <, >, - in each line.
> Allow paths to use only the left-to-right direction. < Allow paths to use only the right-to-left direction. - Disallow paths to use this reaction.
For instance, the following three lines would constrain the reactions R00199, R00200 and R00206 involving PEP -> Pyr so that no edge Pyr -> PEP appears in results because of these reactions. Note that a KEGG reaction file needs to be examined to determine the correct reaction direction - in this case the three reactions have been specified in KEGG in Pyr <- PEP direction, hence the < constraint. To completely forbid a reaction, use the "-" constraint.
R00199 < R00200 < R00206 <
ReTrace generates html results file for the query named according to source and target metabolite identifiers. For instance, for a query from Acetyl-CoA to Lysine, a main html result file named pathways-C00024-to-C00047.html would be generated in the directory specified with the option -o. In addition, a html file is generated for each pathway found. These are accessible from the main html result file, which reports for each summary information including composite mapping, ZO, average reaction score, number of RPAIRs and reactions utilized and number of reactions appearing on the pathways having zero or low reaction score. Currently, low score threshold can be only specified by changing a constant in the source file htmlexport.py.
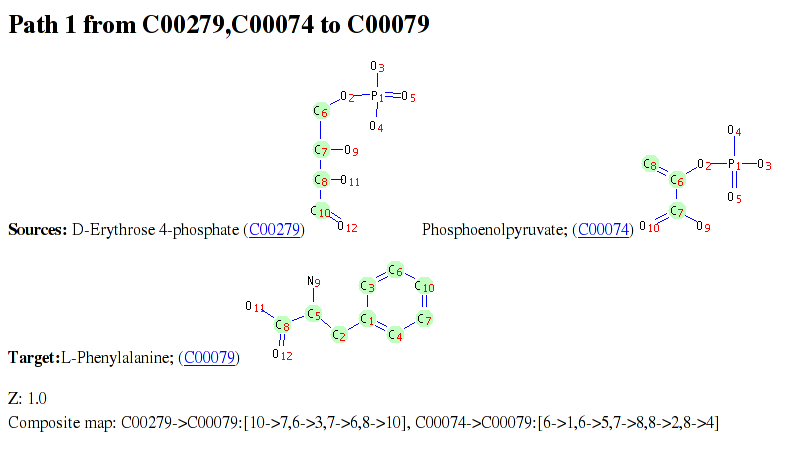
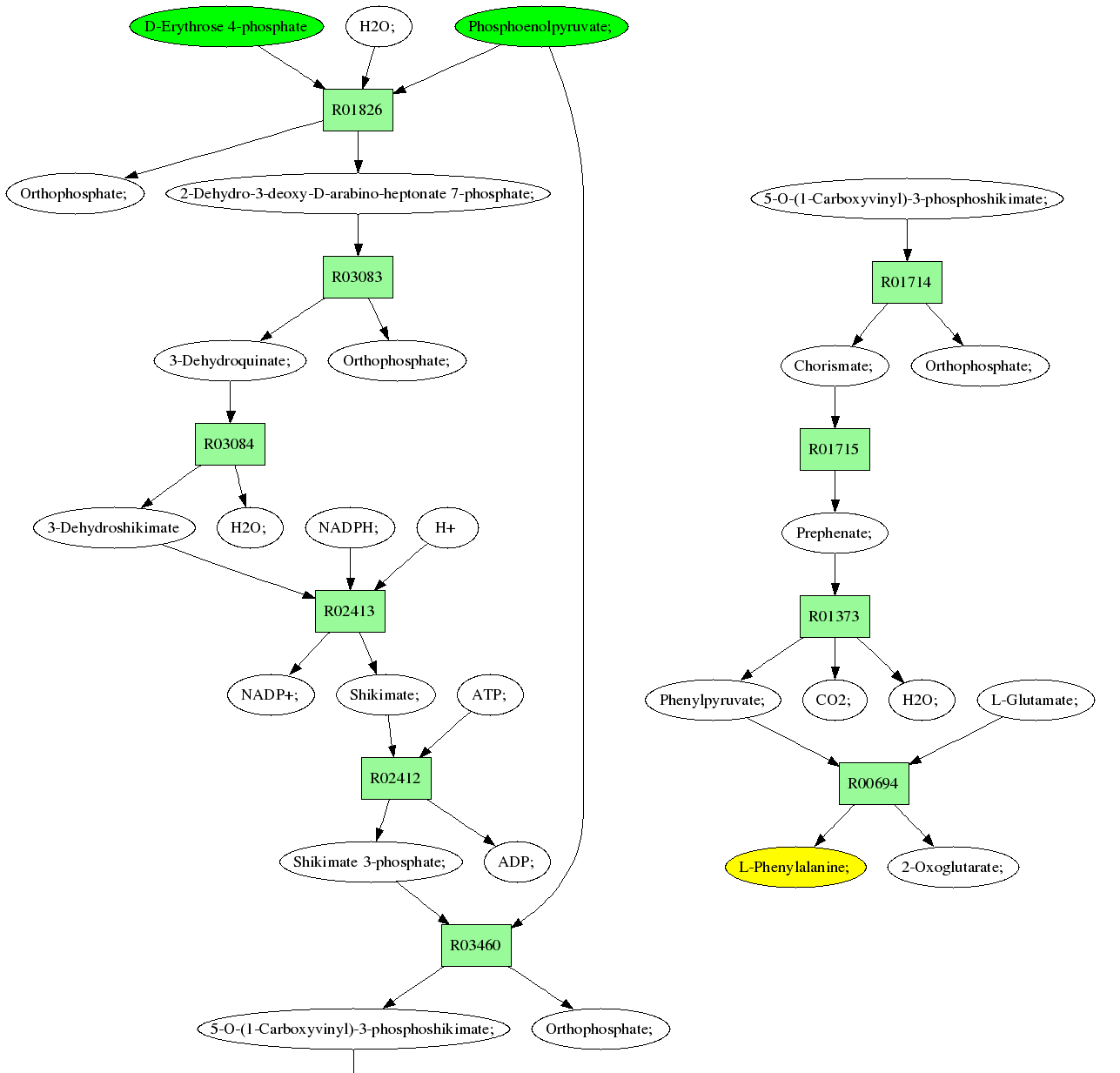
The above figure shows an excerpt from a pathway result file for a query from E4P and PEP to Phe. In addition to the molecule structures with transferred atoms indicated, the pathway result file contains a table detailing each RPAIR and reaction associated with the pathway. Finally, if Graphviz has been available during ReTrace execution as discussed above, a pathway diagram is shown (example figure). In diagram, source and target metabolites are colored green and yellow, respectively. If reaction scores have been provided, reactions with zero score and low score are colored red and blue, respectively. Reactions with scores above threshold are colored green.
For convience, identifiers in the result tables and pathway diagrams are hyperlinked to appropriate KEGG web site pages for easier interpretation of results.
You need to have the following software installed and available in path so they can be invoked from command line.
python retrace.pyYou should see a basic summary of command line options.
{kind=link}