 The basic procedure for comparing two orthologous sequences is
simple as follows
The basic procedure for comparing two orthologous sequences is
simple as follows
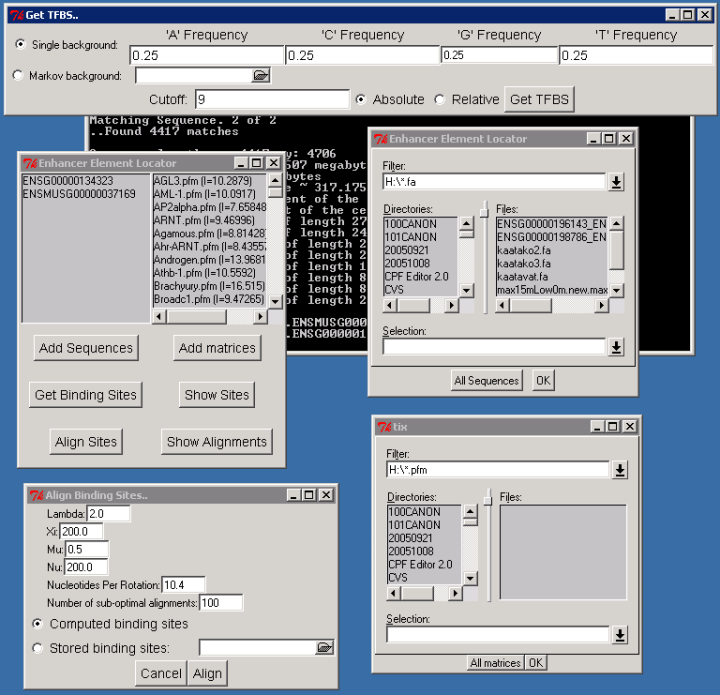
This web page tries to provide some guidance in the usage of the EEL sofware. These instructions refer to the TclTK/Tix user interface as seen in the image below. This interface is available on Windows and on most Unix systems with Xwindowing environment. The important exception is the MacOS X which has it's own MacEEL.
You can try out EEL with example sequences and binding site matrices.
The basic procedure for comparing two orthologous sequences is
simple as follows
Removing a sequence or matrix from the user interface is done by double clicking its name.
You can look and save the list of putative binding sites (mostly junk) by clicking "Show sites" and the list of predicted enhancer elements by clicking "Show alignments". If the DNA sequence is added to the program, the alignment output will be nice "DNA alignment like". If not (you have either removed the sequences or aligned a stored list of putative binding sites), the output will be just numbers.
### lambda=2 mu=0.5 nu=200 xi=200 Nucleotides per rotation=10.4 ### D[ENSMUSG00000037169][ENSG00000134323] Note! First nucleotide at position 1 (one) and binding site at zero!First lines give out the used parameter values and the names of the used sequences. Here the sequence names are ENSMUSG00000037169 and ENSG00000134323
### Alignment No 1 ### D[1762][2445]=38.72 S8.pfm (18430,18434) <=> (24317,24321) + D[1766][2449]=75.43 Tal1beta-E47S.pfm (18462,18473) <=> (24349,24360) + D[1772][2451]=92.13 Pax6.pfm (18527,18540) <=> (24414,24427) + D[1774][2455]=111.26 Broadc4.pfm (18583,18593) <=> (24470,24480) - D[1777][2456]=151.13 SOX17.pfm (18603,18611) <=> (24490,24498) + D[1779][2461]=196.33 NF-kB.pfm (18620,18629) <=> (24507,24516) + D[1780][2464]=232.21 Thing1-E47.pfm (18647,18656) <=> (24534,24543) - D[1787][2470]=216.88 S8.pfm (18763,18767) <=> (24649,24653) - D[1788][2471]=254.45 Myf.pfm (18776,18787) <=> (24662,24673) +On the fist line is the rank of the alignment (the best local alignment on this region).
Sequence 1: ENSMUSG00000037169 Sequence 2: ENSG00000134323 18421 : g-aagagaacAATTAagttc-ttttccctagg-catgtgtggaaTCAACATCTGGAtgcc 24308 : gcaa-agagcAATTA-gcttcttttct-tgggacacatatagaaTCAACATCTGGA-gac 18478 : t-cgagcctgaggcccattacaagtagcaaaagaatttggacaggatgcaTTTAATCTTG 24364 : cacgagcctgagacccatttcaagtagcaaaagaatttggacaggatgcaTTTAATCTTG 18537 : AGTTaatggtacagtgctccgccaaga-aaaacctctcagcttcaacATATTTTACTTag 24424 : AGTTaatggtacaaagctcagccaagataaaat-tctcagcctcaacATATTTTATTTtg 18596 : aaaaactGTTATTGTCaggtacagGGGAAATTCTtcctccctttggctgctCTGCCAGAT 24483 : aaaaaatGTTATTGTCaggtacaaGGGAAATTCCtcctaacttagggggctCTGCCAGAT 18656 : Gacatagttactgcaggggaccact-gacctggtgtgttatctctttcatctgagaaagt 24543 : Gacataagcactggaggg-accattcgtc-tggtgtgttatctttttcatctaagtaggt 18715 : ctctcccccgcagactcatctcccccaaacctggccatgctccctgtgTAATTctagcct 24601 : ctttcctccaccaactcatctctcaaaaagctggccacagttcctaagTAATTctatccc 18775 : cTGACAGCTGCAGgagaaggaag- 24661 : cTGACAGCTGCAGga-aagaaaatFinally there is the DNA like alignment. The EEL algorithm proper have aligned only the sequences with upper case letters. The lower case nucleotides are aligned just for illustrative purposes.
You can use eel on command line by giving terminal commands preceeded by '-' on the command line. The commands are executed from left to right.
In the terminal mode, command 'help' provides the list of available commands and their short introduction.
'-?', '-h', '-help'
Arguments: none
prints this help
'-about'
Arguments: none
Prints Information about the program
'-addMatrix', '-am'
Arguments: filelist
reads matrices from files
'-addSequence', '-as'
Arguments: filelist
reads sequences from files
'-addSingleSequence', '-ass'
Arguments: filelist
Gzipped and Fasta formated sequence files. One sequence in file.
'-align'
Arguments: [filename[,num_of_align,[lambda[,xi[,mu[,nu,[,nuc_per_rotation]]]]]]]
aligns the computed BS or optional the BS from a gff file
filename specifies a file in gff format is you want to be aligned
num_of_align specifies how many alignments you want. (Default 3)
lambda Bonus factor for hit (Default 2)
xi Penalty factor for rotation (Default 1.0)
mu Penalty factor for average distance between sites (Default 0.5)
nu Penalty factor for distance difference between sites (Default 1.0)
nuc_per_rotation specifies how many nucletides there are per rotation. (Default 10.4)
If you want to skip a argument just write '.' for it.
If you use '.' as filename the local data are aligned.
'-cd'
Arguments: [path]
Change or display the current working directory.
'-dir'
Arguments: [path/pattern]
List files matching the given pattern. Defaults to "*" in current working directory.
'-getTFBS'
Arguments: [bound]
computes the scores of all matrices and all sequences which are
better than bound*maxscore. maxscore is the highest reachable
score of the actual matrix with respect to the background
The default value for bound is 0.1
'-getTFBSabsolute'
Arguments: [cutoff]
computes the scores of all matrices and all sequences which are
better than cutoff.
The default value for cutoff is 9.0
'-more'
Arguments: [number of alignments]
prints more alignments from previously run alignment matrix
'-no-gui'
Arguments: none
Gives command line interface
'-pm', '-printMatrices'
Arguments: none
prints the matrices
'-pmw', '-printMatrixWeights'
Arguments: none
prints the matrix weights (with background)
'-printSeqNames', '-ps'
Arguments: none
prints the names of the sequences
'-q', '-quit'
Arguments: none
to exit the program
'-removeMatrix', '-rm'
Arguments: Matrixnumber
removes a matrix
'-removeSequence', '-rs'
Arguments: Sequencename
removes a sequence
'-reset'
Arguments: none
removes all matrices and sequences
'-resetMatrices', '-resm'
Arguments: none
removes all matrices
'-resetSequences', '-ress'
Arguments: none
removes all sequences
'-sa', '-showalign'
Arguments: none
prints the computed alignment to stdout
'-saveMarkovBackground'
Arguments: filename
Name of the file where to store the background model.
'-savealign'
Arguments: [filename]
saves the alignment to disk
The default filename is 'eel_[Date+Time].align'
e.g. eel_2003_9_16_11_48.align
'-savealignAnchor'
Arguments: [filename]
saves the alignment to disk in Anchor format for DIALIGN
The default filename is 'eel_[Date+Time]_align.anc'
e.g. eel_2003_9_16_11_48_align.anc
'-savealignGFF'
Arguments: [filename]
saves the alignment to disk in GFF format
The default filename is 'eel_[Date+Time]_align.gff'
e.g. eel_2003_9_16_11_48_align.gff
'-savematch'
Arguments: [filename]
saves the results of the matching in gff format
See http://www.sanger.ac.uk/Software/formats/GFF/
The default filename is 'eel_[Date+Time].gff'
e.g. eel_2003_8_27_15_48.gff
'-setBGfreq'
Arguments: A C G T
Background nucleotide frequencies. Removes markov background.
'-setMarkovBG'
Arguments: bgSampleSequence [order]
Background sample sequence and order of the model or saved background file.
'-setpseudocount'
Arguments: [pseudocount]
Set the amount of pseudocounts on matricies. Default 1.0
'-showmatch', '-sm'
Arguments: none
prints the computed scores to stdout