1. De Bruijn graphs and kmer sets
- GGCAT: Extremely-fast construction and querying of compacted and colored de Bruijn graphs
- matchtigs & eulertigs: Minimum plain-text representation of kmer sets / spectrum preserving string sets
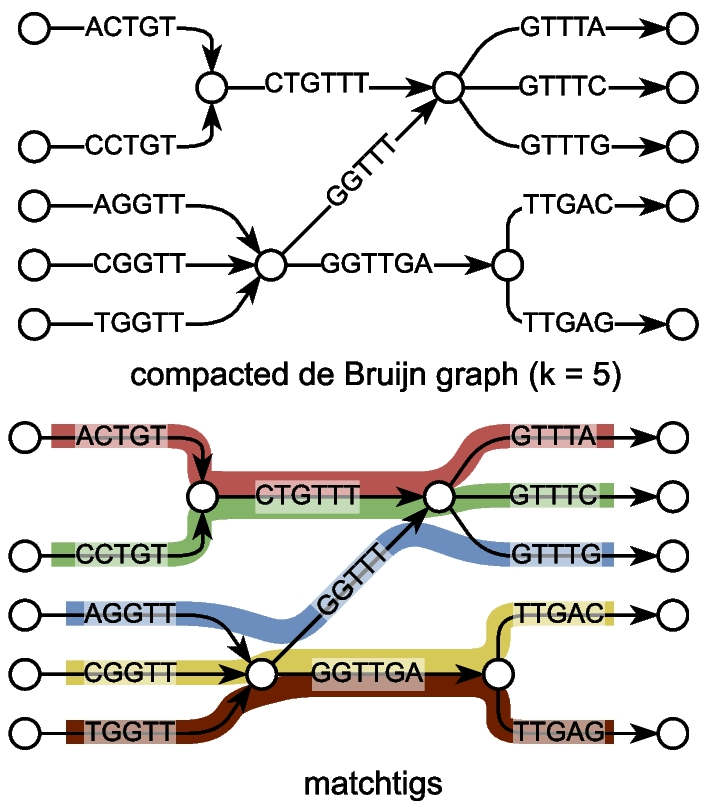
1. De Bruijn graphs and kmer sets
|
|
2. Fast solvers for flow decomposition problems via Integer Linear Programming
|
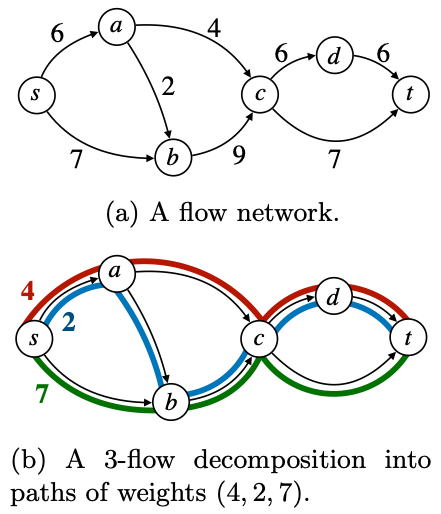
|
3. Alignment to pangenomes
|
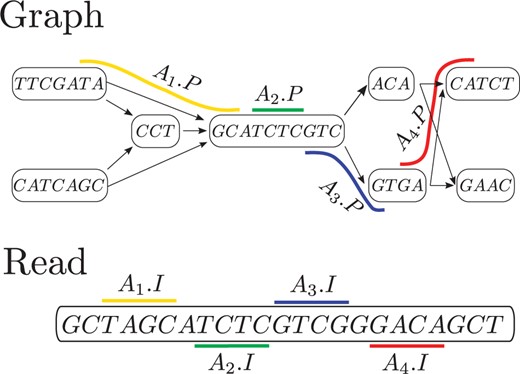
|
4. Super-unitigs for genome assembly
|
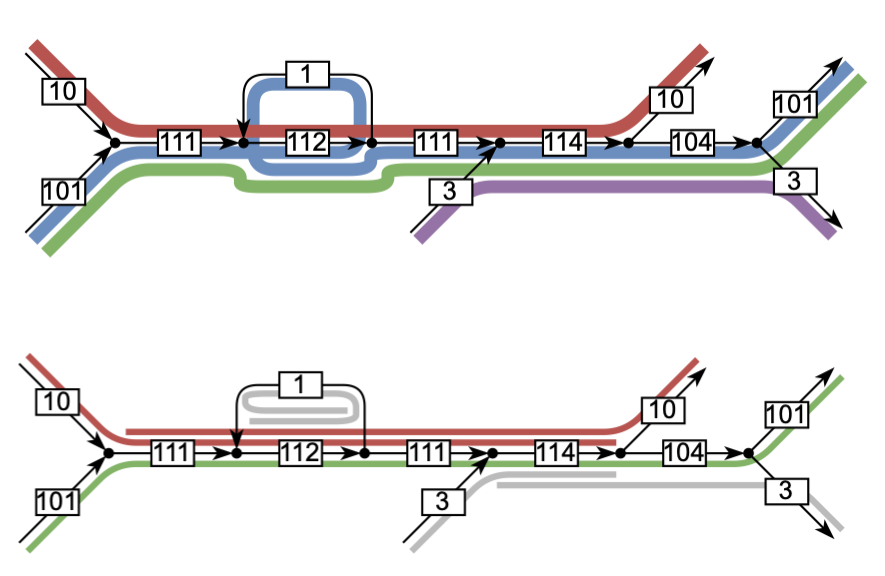
|
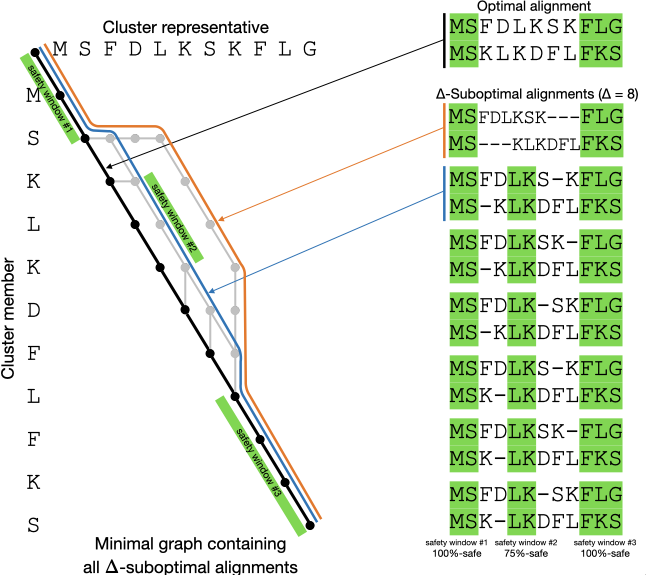
5. Protein and RNA alignment
|
|