
The Discovering Network Structures (DiNS) project is a research collaboration between several prominent researchers at Algodan / HIIT working on methods for inferring networks from large amounts of quantitative data.
Note that there is an internal wiki for discussion among DiNS researchers.
Network-like structures are numerous in various domains. For example, the molecular level processes of a living organism can be modelled by using gene regulatory networks and metabolic networks. The human society is full of different social networks. The Internet is a huge network, with a plethora of subnetworks. New computational methods are needed for finding the structure of such networks and for understanding their dynamic behaviour. As explicit knowledge of the networks is typically missing, we have to resort to computational techniques that reconstruct networks from data. As growing amounts of data describing such networks are available, we see improved possibilities for significant progress on this very demanding area.
|
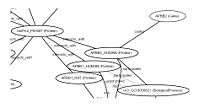
BiominePublic biological databases contain huge amounts of rich data, such as annotated sequences, proteins, domains, and orthology groups, genes and gene expressions, gene and protein interactions, scientific articles, and ontologies. The Biomine project develops methods for the analysis of such collections of data. Representative publications Link discovery in graphs derived from biological databases Finding Reliable Subgraphs from Large Probabilistic Graphs Demo The Biomine prototype search engine for biological data integrates data from several publicly available biological databases with the aim of aiding explorative discovery of connections between biological entities, such as genes and phenotypes: http://biomine.cs.helsinki.fi/
|

|
SeriationThe problem in seriation is finding an ordering (total or partial order) among entities. For instance, in paleontology, the data may consist of a collection of fossil sites, a set of taxa, and the presence/absence information for all taxa, and the problem is to find a good ordering for the sites. Representative publications Seriation in Paleontological Data Using Markov Chain Monte Carlo Methods Software Software for seriation in paleontology data
|

|
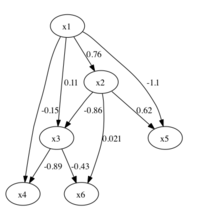
Learning discrete-valued Bayesian networksSeveral DiNS-researchers have worked on the topic of learning Bayesian networks for discrete-valued random variables. Both Bayesian approaches and methods based on the Minimum Description Length principle have been investigated. Representative publications Exact Bayesian structure discovery in Bayesian networks Advances in exact Bayesian structure discovery in Bayesian networks A Simple Approach for Finding the Globally Optimal Bayesian Network Structure On Sensitivity of the MAP Bayesian Network Structure to the Equivalent Sample Size Parameter Factorized NML Criterion for Learning Bayesian Network Structures Software and demos The computer program REBEL is available from Mikko Koivisto. B-course is a web-based data analysis tool for Bayesian modeling, in particular multi-variate dependence and causal modeling and classification, hosted by the CoSCo group. Bene is a software for exact structure learning hosted by the CoSCo group. The program source code for Linux and Windows can be accessed from the demo page. |
|
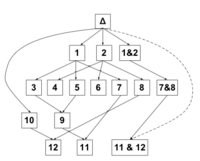
Learning continuous-valued linear causal networksIn the LiNGAM project, we develop methods for identifying data-generating processes with a DAG structure, where the observed variables are continuous-valued and all the mechanisms are linear. Representative publications A linear, non-gaussian acyclic model for causal discovery Estimation of causal effects using linear non-gaussian causal models with hidden variables Software We distribute a LiNGAM matlab package for performing the basic LiNGAM analysis.
|
|
Modeling metabolic networksIn this project, computational methods for reconstructing metabolic networks are developed. The methods utilize optimization techniques and graph traversal algorithms to discover a set of biochemical reactions that is most likely catalyzed by the enzymatic genes of the target organism. An important aspect of the developed methods is that they generate networks which are structurally consistent (gapless). Representative publications An analytic and systematic framework for estimating metabolic flux ratios from 13C tracer experiments A computational method for reconstructing gapless metabolic networks
|
|
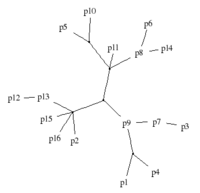
Computer-assisted stemmatologyStemmatology (a.k.a. stemmatics) studies relations among different variants of a document that have been gradually built from an original text by copying and modifying earlier versions. The aim of such study is to reconstruct the family tree (causal graph) of the variants. Representative publications A Compression-Based Method for Stemmatic Analysis Evaluating methods for
computer-assisted stemmatology using artificial benchmark data sets |